
Một số bất thường nhiễm sắc thể thường gặp ở trẻ sơ sinh
Bất thường nhiễm sắc thể (NST) là thay đổi số lượng hay cấu trúc NST thường gắn với các dị tật bẩm sinh và các bất thường khác. Tỉ lệ bất thường NST khoảng 5/1000 trẻ sơ sinh sống (0.5%).Phần lớn bất thường NST là bất thường mới xuất hiện lần đầu (nghĩa là, các thành viên khác trong gia đình không có bất thường này).
Bất thường NST thường được phân loại dựa trên số lượng, cấu trúc hay nội dung. Bệnh lý có thể liên quan tới NST thường hay NST giới tính; bệnh do bất thường NST thường hay nặng hơn so với những bệnh lý do bất thường NST giới tính.

Bất thường số lượng là sự sai lệch số lượng NST hoàn chỉnh (nguyên bội). Ví dụ của thể dị bội (aneuploidy) bao gồm thể tam bội 21 (Hội chứng Down), thể tam bội 18, thể tam bội 13, hội chứng Klinefelter (47, XXY), và hội chứng Turner (45,X).
Bất thường cấu trúc là kết quả của sự đứt gãy và sắp xếp lại cấu trúc NST. Các khả năng có thể bao gồm chuyển đoạn không cân bằng, mất đoạn (nhỏ), lặp đoạn, đảo đoạn, và NST đều (isochromosome). Ví dụ của bệnh lý do bất thường cấu trúc NST gồm hội chứng mất đoạn nhỏ 22q11, hội chứng Cri du chat (xóa đoạn 5p), và u Wilms với dị tật không có mống mắt (xóa đoạn 11p). Hội chứng Prader – Willi thường gắn với chứng mất đoạn (nhỏ) trên NST 15.
MỤC LỤC
1. Bất thường số lượng NST (đa bội)
2. Bất thường cấu trúc nhiễm sắc thể
1. Bất thường số lượng NST (đa bội)
Thể tam bội (trisomy) đề cập đến khái niệm có ba hay nhiều hơn hai NST bình thường – bản sao của NST đặc hiệu xuất hiện ở trong các tế bào của một người. Thể tam bội này hầu như luôn xuất hiện do lỗi trong quá trình giảm phân gọi là hiện tượng không phân ly trong noãn bào hoặc tinh bào. Chỉ một số thể tam bội được phát hiện ở trẻ sơ sinh, những thể khác chỉ được thấy ở bào thai bị sảy tự nhiên.
1.1 Hội chứng Down (DS) là thể tam bội thường gặp nhất.
Karyotype phổ biến nhất của hội chứng Down là trisomy 21, mặc dù một số trường hợp có thể do chuyển đoạn hoặc, cực hiếm là thể khảm.
- Thể tam bội. 95% trẻ em bị Down có 47 nhiễm sắc thể, với 3 NST 21. Trisomy 21 xảy ra ở 1/700 trẻ sơ sinh.
+ Nguy cơ sinh con có 3 NST 21 tăng cao cùng với tuổi người mẹ. Nguy cơ tăng rõ sau 35 tuổi. Tuy nhiên, phần lớn trẻ có trisomy 21 lại được sinh ra bởi phụ nữ nhỏ hơn 35 tuổi, bởi vì phần lớn phụ nữ sinh con trước năm 35 tuổi. Lí do làm tăng tỉ lệ trisomy 21 ở bào thai của phụ nữ lớn tuổi vẫn chưa được hiểu rõ. NST thừa có nguồn gốc từ người cha chỉ chiếm một phần nhỏ trong các trường hợp.
+ Theo quan sát thực tế cho thấy nguy cơ tái xuất hiện cho cha mẹ có trẻ mắc thể tam bội 21 là ~1% (trừ khi nguy cơ cao hơn liên quan đến cha mẹ lớn tuổi).
Chuyển đoạn. 4% trẻ mắc bệnh Down có 46 NST và một chuyển đoạn NST 21 thứ ba tới NST khác – thường là NST số 13, 14, 15, 21 hoặc 22.
+ ¾ các trường hợp chuyển đoạn trong hội chứng Down là mới phát sinh (nghĩa là: không có tính chất gia đình).
+ ¼ các trường hợp chuyển đoạn có tính chất gia đình, nghĩa là bố hoặc mẹ có chuyển đoạn cân bằng liên quan tới một NST 21 và NST khác. Trong các trường hợp này, nguy cơ tái phát theo quan sát thực nghiệm có thể cao tới 15% cho các lần mang thai tiếp theo (phụ thuộc vào NST nào liên quan và giới tính của người mang chuyển đoạn cân bằng). Trong trường hợp hiếm của chuyển đoạn NST 21 có tính chất gia đình, nguy cơ tái phát là 100%.
- Thể khảm. 1% trẻ bị Down là thể khảm, với một số tế bào có 46 NST (2 NST 21), và một số tế bào có 47 NST với 3 NST 21. Thể khảm là do lỗi xảy ra trong quá trình phân bào ở thời kì đầu phát triển phôi hoặc mất NST 21 số ba ở trong lớp tế bào đầu. Trẻ bị hội chứng Down thể khảm có thể có biểu hiện bệnh nhẹ hơn với trẻ bị trisomy 21 ở trong mọi tế bào.
- Đặc điểm lâm sàng. Trẻ bị hội chứng Down có ngoại hình đặc trưng được xác định bởi đặc điểm bất thường hình thái, ngoài ra còn có dị tật chức năng và cấu trúc đặc trưng ở các cơ quan khác. Dị tật tim, đặc biệt là dị tật gối nội mạc tim và dị tật vách tim, được phát hiện ở gần 50% bệnh nhân hội chứng Down.
- Tiên lượng: Với sự phát triển của các biện pháp điều trị y khoa, giáo dục và hướng nghiệp, những người mắc hội chứng Down có thể sống tới tuổi trưởng thành. Bởi vậy, các vấn đề liên quan tới nghề nghiệp, bảo mật tài chính, chăm sóc sức khỏe, và điều kiện sống cần phải được lưu ý. Những người bị Down đang có được cuộc sống bán độc lập.
1.2 Trisomy 13
Khoảng 1/4000 tới 1/10,000 trẻ sơ sinh có thể tam bội 13.
- Các dạng đột biến thường gặp là:
+ Thể tam bội. 75% các trường hợp có thể tam bội 13 gây ra bởi sự tăng thêm mỗi NST 13 - kết quả của sự không phân ly trong giảm phân. Có mối liên quan giữa sự xuất hiện của thể tam bội 13 và tuổi sinh sản, mặc dù nó không mạnh như trong thể tam bội 21.
+ Chuyển đoạn. 20% các trường hợp thể tam bội 13 là kết quả của sự chuyển đoạn.
¾ các trường hợp này là mới phát sinh (de novo).
¼ các trường hợp bị gây ra bởi sự chuyển đoạn tuyến gia đình liên quan tới NST 13, và trong những trường hợp này nguy cơ tái phát theo quan sát thực nghiệm có thể cao tới 14% ở các lần mang thai sau.


+ Thể khảm. 5% các trường hợp mắc thể tam bội 13 là thể khảm với các dòng tế bào 46 NST thường và 47 NST với thêm một NST 13.
- Đặc tính lâm sàng thể tam bội 13 được liệt kê ở Bảng 1.
- Tiên lượng cho bệnh nhân mắc hội chứng này cực kì kém: 50% bệnh nhân tử vong trước 1 tháng tuổi, 70% tử vong trước 6 tháng tuổi và 90 tử vong trước 1 năm tuổi.
1.3 Thể tam bội 18
Xảy ra ở 1/8000 trẻ sơ sinh.Tuổi sinh sản có liên quan tới sự xuất hiện của thể tam bội 18, nhưng ít được ghi nhận hơn so với thể tam bội 21 và 13.
- Các loại dị tật. Thể tam bội 18 hiếm khi gây ra bởi chuyển đoạn nhiễm sắc thể.
+ 90% các trường hợp mắc thể tam bội 18 là kết quả của giảm phân.
+ 10% các trường hợp mắc thể tam bội 18 là thể khảm – dù gây ra bởi giảm phân hậu hợp tử hay do mất NST 18 số 3 ở trong dòng tế bào từ phôi.
- Đặc điểm lâm sàng của trisomy 18 được liệt kê trong Bảng 1. Tính đa dạng của triệu chứng và các dị tật hình thái rất nhỏ có thể làm bệnh khó chẩn đoán.
- Tiên lượng cho bệnh nhân trisomy 18 kém: 30% bệnh nhân tử vong trong giai đoạn sơ sinh, 90% tử vong trước 1 tuổi.
1.4 Bất thường NST giới tính
Do bất thường về số lượng hay cấu trúc của NST X hay Y.
1.4.1. Hội chứng Turner: tỉ lệ mắc khoảng 1/3000 trẻ sơ sinh nữ.
- Các dạng bất thường. Ở hội chứng Turner, NST X thứ hai có thể bị mất hay bất thường. Kiểu bất thường NST khác nhau gây ra các kiểu hình khác nhau của hội chứng Turner.
+ 55% bệnh nhân hội chứng Turner, có karyotype là 45,X.
+ 25% bệnh nhân hội chứng Turner có cấu trúc của một trong hai NST X bị biến đổi. Bất thường cấu trúc thường là mất đoạn NST hoặc lặp đoạn cánh dài hay cánh ngắn của NST trên cánh còn lại (gọi là NST đều).
+ 15% bệnh nhân, có tồn tại hai hay nhiều dòng tế bào được gọi là thể khảm, một dòng tế bào là 45,X và dòng tế bào còn lại là 46,XX hay 46,XY. Có thể có dòng tế bào thứ ba, thường gặp là karyotype 45,X/46,XX/47,XXX.
Nguy cơ tái xuất hiện của cha mẹ có con gái mắc hội chứng Turner bằng nguy cơ của dân số chung (nghĩa là: 1/2500 bé gái sống, hoặc chính xác hơn, 1/5000 trẻ sơ sinh sống).
Đặc điểm lâm sàng của hội chứng Turner có thể được thấy ngay từ khi sinh ra, mặc dù nhiều bé gái không được chẩn đoán cho tới khi dậy thì.
+ Dị tật hình thái bao gồm phù bạch huyết ở tay và chân bẩm sinh, ngực hình khiên, thừa da cổ, cẳng tay cong ngoài (tăng góc vẹo), tầm vóc thấp (người trưởng thành cao trung bình 135 cm [53 inches]), và nhiều nốt ruồi sắc tố.
+ Bất thường về chức năng và cấu trúc tuyến sinh dục: chủ yếu gây vô kinh và không phát triển dậy thì do không có hormon buồng trứng. U tế bào mầm (khối u ở buồng trứng có các tế bào mang NST Y) có thể gặp ở những bệnh nhân hội chứng Turner thể khảm mang dòng tế bào có một NST Y.
+ Dị tật thận, xuất hiện ở khoảng 40% người bệnh, gồm niệu quản đôi và thận móng ngựa.
+ Dị tật tim xảy ra ở 20% bệnh nhân. Dị tật gồm hẹp van động mạch chủ, van động mạch chủ 2 lá, và hẹp eo động mạch chủ. Phình động mạch chủ ở người trẻ tuổi có thể gây ra biến chứng đe dọa tính mạng.
+ Viêm tuyến giáp tự miễn thường gặp.
+ Trí tuệ thường bình thường. Tuy nhiên, rối loạn khả năng học tập ở một số lĩnh vực thường xảy ra, đặc biệt trong môn hình học không gian.
Chẩn đoán hội chứng Turner dựa trên kết quả NST đồ. Tiêu chuẩn là phân tích 30 tế bào, nên có thể phát hiện được thể khảm ở mức độ thấp.
Tiên lượng phụ thuộc vào dạng bất thường NST và mức độ nghiêm trọng của các dị tật. Tuổi thọ có thể bình thường trong phần lớn các trường hợp. Điều trị hormone tăng trưởng tái tổ hợp có thể giúp người nữ mắc hội chứng Turner đạt được chiều cao trưởng thành.
Hội chứng Klinefelter: Tỉ lệ mắc khoảng 1/800 trẻ nam sơ sinh sống, do thừa 1 NST X.
- Các dạng bất thường
+ 80% bệnh nhân mắc hội chứng Klinefelter có karyotype 47,XXY.
+ 20% bệnh nhân thể khảm với một dòng tế bào có karyotyple 47,XXY.
Nguy cơ tái xuất hiện của hội chứng Klinefelter giống như tỉ lệ mắc của dân số chung (nghĩa là: 1/1600 trẻ sơ sinh sống sót).
+ Đặc điểm lâm sàng rất đa dạng và không đặc hiệu.
+ Bé trai thường cao, gầy, chiều dài sải tay dài hơn nhiều so với chiều
+ Tinh hoàn nhỏ
+ Nồng độ testosterone trong huyết thanh thường thấp.
+ Ở tuổi dậy thì, trẻ trai nam hoá không hoàn toàn
+ Vú to ở nam giới là đặc điểm thường thấy, có thể làm tăng tỉ lệ mắc ung thư vú ở nam 47, XXY.
+ Chậm phát triển tâm thần từ nhẹ - trung bình
+ Rối loạn hành vi và tính cách ít gặp
2. Bất thường cấu trúc nhiễm sắc thể
Phần lớn bất thường cấu trúc NST thường gắn với các kiểu hình rất đặc trưng; tuy nhiên, một số lại không. Trong một số trường hợp, phân tích NST của cha mẹ là quan trọng trong việc xác định sự tương quan lâm sàng của bất thường nhỏ trên NST (vd: vùng cận đầu telomera). Một số khác biệt về NST có tính chất gia đình và không có ý nghĩa lâm sàng.
2.1 Mất đoạn/ lặp đoạn một phần
Các dạng bất thường
- Một số hội chứng có thể do mất đoạn tận cùng của NST (terminal deletion), hay do mất vùng giàu gen đầu tận cùng NST (subtelomeric chromosome deletion), hoặc mất vật chất di truyền phần giữa hoặc bên trong của NST (mất đoạn giữa – interstitial deletion). Một số mất đoạn thường xảy ra và dễ nhận biết, như mất đoạn ở vị trí NST 22q11 (xem IV D 1 c [1]).Lặp đoạn cũng gây ra những hội chứng đã được biết đến (lặp đoạn 22q11 gây ra hội chứng cat-eye).
- Mặc dù phần lớn mất đoạn NST đều là bất thường mới (de novo), mất đoạn tận cùng (và rất hiếm khi mất đoạn giữa) có thể gặp ở những bệnh nhân mang NST chuyển đoạn không cân bằng (vật chất di truyền chuyển đổi không cân bằng giữa hai nhiễm sắc thể) từ người cha hoặc mẹ mà có chuyển đoạn cân bằng (vật liệu chuyển đổi cân bằng giữa hai nhiễm sắc thể). Trong các trường hợp mất đoạn, cần phải phân tích NST ở cả cha và mẹ để loại trừ trường hợp chuyển đoạn cân bằng ở cha mẹ.
- Tiền sử sảy thai liên tiếp có thể gặp ở cha mẹ mang chuyển đoạn NST cân bằng bởi vì các thành viên khác trong gia đình cũng có thể mang chuyển đoạn.
2.2 Hội chứng xóa gen liền kề đề cập đến kiểu hình liên quan tới sự vắng mặt một số gen gần đó.
- Những bất thường này gây ra những bệnh lý có thể nhận biết được nhưng đa dạng, biểu hiện triệu chứng phong phú.
- Trong một số trường hợp, mất đoạn nhỏ có thể được quan sát thấy bằng phân tích NST với độ phân giải cao; ngoài ra thì không quan sát được. Trong những trường hợp này, rất cần kĩ thuật FISH để phát hiện mất đoạn.
- Một ví dụ của hội chứng gen liền kề là hội chứng Williams, do mất đoạn nhỏ trên nhánh dài của NST 7, bao gồm cả gen elastin.
+ Bệnh nhân mất đoạn gen elastin đơn thuần chỉ có dị tật tim bẩm sinh, điển hình là hẹp trên van động mạch chủ.
+ Bệnh nhân thiếu thêm các gen liền kề sẽ mắc hội chứng Williams, bị thiểu năng trí tuệ nhẹ, khuôn mặt đặc trưng, rối loạn hành vi (―tính cách thân thiện‖), canxi trong máu cao, và khớp lỏng lẻo, và hẹp trên van động mạch chủ.
+ Những đặc điểm trên được cho là do mất các gen liền kề.
2.3 Mất đoạn tận cùng (Terminal deletions)
Trẻ em bị mất đoạn tận cùng NST thường chậm lớn, bị chậm phát triển tinh thần, bất thường hình thái, và nhiều dị tật khác, dù các kiểu hình rất đa dạng.
Ví dụ các bệnh lý gây ra bởi mất đoạn nhiễm sắc thể
+ Tỉ lệ mắc hội chứng mất 22q11 là 1/4000 trong dân số. Trong một số trường hợp, mất đoạn được phát hiện khi phân tích NST thường quy, nhưng nhiều trường hợp lại chỉ có thể phát hiện vị trí 22q11bằng kĩ thuật FISH.
+ Hội chứng mất đoạn nhỏ đã được chứng minh gây ra hội chứng DiGeorge, hội chứng dị tật vòm – tim – mặt (velocardiofacial), hội chứng dị tật thân chung động mạch tim – dị tật mặt, và một số giống như dị tật thân chung động mạch tim đơn độc.
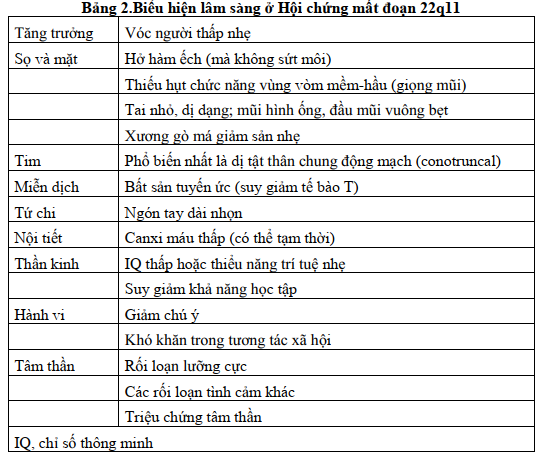
+ Bệnh nhân bị mất đoạn có biểu hiện rất đa dạng. Biểu hiện lâm sàng được mô tả ở Bảng 2.
Một số trẻ sơ sinh bị dị tật tim bẩm sinh (phần lớn là thân chung động mạch), hở hàm ếch, và/hoặc suy giảm miễn dịch nghiêm trọng. Hơn một nửa bệnh nhân mất đoạn 22q11có gián đoạn cung động mạch chủ típ B.
Cha mẹ của những trẻ bị bệnh cũng nên xét nghiệm bởi vì các nghiên cứu cho thấy có 10% tỷ lệ mất đoạn có tính chất gia đình, đôi khi cha mẹ có biểu hiện bình thường.
Hội chứng Cri du chat gặp ở 1/50,000 trẻ sơ sinh.
+ Bất thường. Mất vật chất di truyền ở đầu tận cùng của nhánh nhánh ngắn NST số 5 (5p-).
+ Đặc điểm lâm sàng
Bệnh nhân có tiếng khóc như mèo.
Thiểu năng trí tuệ nặng và bất thường thần kinh trung ương là triệu chứng phổ biến.
Dị tật tim bẩm sinh và dị tật thị giác thường gặp (vd: đục thủy tinh thể, teo thần kinh thị giác).
+ Tiên lượng. Bệnh nhân có thể sống tới tuổi trưởng thành.
2.4 Thể tam bội một phần
Thể tam bội một phần xảy ra khi thêm một phần nhiễm sắc thể.
- Đoạn NST thêm có thể gắn vào đầu tận cùng của cánh dài hay cánh ngắn của nhiễm sắc thể, hoặc nó sẽ thêm vào đoạn giữa NST bình thường.
- Thường thì một đoạn nhỏ của NST thêm vào, có thể có hoặc không có tâm động, được tìm thấy trên NST đồ. Đó là marker nhiễm sắc thể.Một số marker NST không ảnh hưởng đến kiểu hình. Khi marker được phát hiện trước sinh có thể chứng minh rằng cha hoặc mẹ cũng có marker này, do đó tiên lượng thường tốt cho thai nhi.
- Đặc điểm lâm sàng. Nguồn gốc của đoạn NST thêm quyết định mức độ ảnh hưởng tới kiểu hình.Khi đoạn NST thêm có nguồn gốc từ NST thường, thì kiểu hình thường là bất thường hình thái, chậm tăng trưởng, chậm phát triển tâm thần và các dị tật khác.
- Chẩn đoán
+ Phân tích NST với độ phân dải cao hay kĩ thuật FISH đôi khi phát hiện nguồn gốc của đoạn NST thêm, đặc biệt nếu đoạn đó lớn.
+ NST đồ của cha mẹ nên thực hiện bởi vì thể tam bội một phần thường do chuyển đoạn cân bằng ở cha mẹ gây ra.
TÀI LIỆU THAM KHẢO
- Gardner RJM. (2011). Chromosome Abnormalities and Genetic Counseling 4th edition. Oxford Monographs on Medical Genetics.
- Hurst HVFJA. (2017) Clinical Genetics and Genomics. 2nd, edition. Oxford Desk Reference.
- Jones KL. (2017). Smith's recognizable patterns of human malformation. 6th, edition.
"Hướng dẫn chẩn đoán và điều trị bệnh trẻ em (cập nhật 2020)", BV Nhi Trung Ương
Anh chị có thể inbox Pan Happy hoặc liên hệ qua hotline zalo 0964821468 để được hướng dẫn dinh dưỡng cho mẹ và bé khỏe nha ạ.










