
Hội chứng DiGeorge ở trẻ nhỏ: Nguyên nhân, triệu chứng và cách điều trị
Hội chứng DiGeorge hay còn gọi là hội chứng mất đoạn 22q11 là một trong những hội chứng mất đoạn nhỏ thường gặp nhất ở người. Tỉ lệ mắc bệnh 1/4000, tương đương giữa nam và nữ.
MỤC LỤC
1. Nguyên nhân

Hội chứng mất đoạn 22q11 là sự mất đoạn từ 1,5 đến 3 Mb trên nhánh dài (q) của nhiễm sắc thể số 22. Đây là kiểu mất đoạn nhiễm sắc thể thường gặp nhất ở người.
Trên lâm sàng có thể có một số hội chứng lâm sàng riêng biệt, chủ yếu là DiGeorge, Shprintzen hoặc Velocardiofacial và Conotruncal. Việc chẩn đoán thường bị trì hoãn từ vài tháng hoặc vài năm, một phần do các bác sĩ chuyên khoa có thể không đánh giá được mối liên hệ di truyền giữa các khuyết tật khác nhau.
Việc mất đoạn có thể tự phát trong 85% các trường hợp hoặc di truyền từ bố mẹ bị bệnh. Bệnh di truyền trội trên nhiễm sắc thể thường, do đó với nguy cơ xuất hiện ở đời con là 50%. Nguy cơ mắc bệnh từ người bố hoặc mẹ bình thường, nhưng mang mất đoạn nhiễm sắc thể ở tế bào noãn hoặc tinh trùng (đột biến tế bào mầm sinh dục) là 1%.
2. Triệu chứng
2.1 Triệu chứng lâm sàng
Đây là một bệnh lý có tổn thương đa cơ quan và đặc điểm về tuổi được coi là triệu chứng đầu tiên. Mức độ nặng của bệnh rất khác nhau giữa các bệnh nhân và các thành viên trong gia đình.
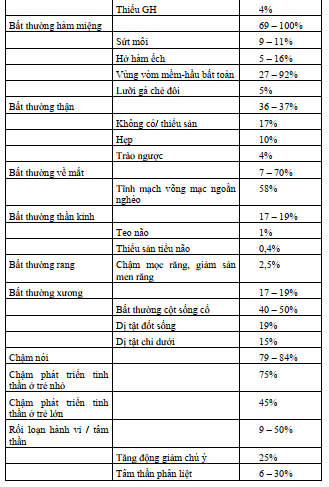
Thay đổi đặc điểm khuôn mặt thường rất tinh tế trong giai đoạn sơ sinh, đôi khi rất khó nhận ra. Thường trẻ có khuôn mặt dài hẹp, mắt hình quả hạnh nhân, mũi củ hành, miệng nhỏ, tai gấp nếp, hộp sọ không cân xứng do dính khớp sọ.
Tim bẩm sinh gặp ở khoảng 50 - 85% các trường hợp. Chủ yếu là tứ chứng Fallot, hẹp động mạch phổi, MAPCA (tuần hoàn bàng hệ chủ phổi) hoặc gián đoạn quai động mạch chủ. Ngoài ra, có thể suy tim sớm do thông liên thất shunt lớn.
Hạ canxi máu gặp ở khoảng 30 đến 60%. Biểu hiện như bồn chồn, co giật, thở rít, xét nghiệm sinh hoá có suy tuyến cận giáp, thường được phát hiện sau sinh, khi phẫu thuật tim, dậy thì hoặc khi mang thai. Men răng yếuvà dễ bị sâu răng.
Rối loạn miễn dịch ảnh hưởng đến đa số các bệnh nhân, nhưng tương đối nhẹ. Chỉ khoảng 1% các bệnh nhân bị suy giảm miễn dịch nghiêm trọng, cần đến ghép tuyến ức. Nhiễm trùng đường hô hấp trên tái diễn thường tăng cùng với khe hở vòm họng. (VPI). Các bệnh tự miễn dịch như viêm khớp dạng thấp vị thành niên, viêm khớp, giảm tế bào máu, bệnh celiac và bệnh lý tuyến giáp.
Bú hay ăn kém là triệu chứng khá phổ biến, khoảng 40%. Nguyên nhân thường gặp bao gồmdị tật vòm miệng (14%), trào ngượcdạ dày - thực quản, khó nuốt (10%) có thể liên quan đến lõm ngực, suy tim, vàchậm phát triển. Bệnh cũng ảnh hưởng đến tăng trưởng. 40% bệnh nhân có chiều cao và cân nặng dưới 3 bách phân vị trong năm đầu tiên. Tuy nhiên trẻ có thể bắt kịp tăng trưởng vào cuối thời thơ ấu với chiều cao dưới trung bình một chút. Tỷ lệ thừa cân và thiếu hụt hormone tăng trưởng tương đương dân số nói chung.
Các vấn đề về ngôn ngữ và giao tiếp gặp ở khoảng ở 90%, đặc trưng bởi, thường khó khăn trong diễn đạt và chậm phát triển ngôn ngữ. Điếc là dođến viêm tai giữa và viêm tai giữa thanh dịch gặp khoảng 75% các trường hợp; 15% bị điếc thần kinh giác quan.Hầu hết trẻ đều có giảm khả năng học tập, chỉ số IQ trung bình khoảng 70.
Các triệu chứng khác có thể gặp bao gồm táo bón và đau chân không rõ nguyên nhân. Vẹo cột sống có ý nghĩa lâm sàng tương đốiphổ biến (18%). Bệnh có thể do bất thường cấu trúc, xuất hiệnsớm hoặc muộn hơn từ 10 đến 12 tuổi, tương tự nhưvẹo cột sống vô căn ở trẻ vị thành niên.
Rối loạn hành vi và tâm thần gặp khoảng 93%. Trong thời thơ ấu, có thể rối loạn phổ tự kỷ, tăng động giảm chú ý, tâm lý hoảng sợ, lo âu, ám ảnh, thụ động và kỹ năng xã hội kém là các triệu chứng phổ biến. Các triệu chứng loạn thần có thể xuất hiện trongthời niên thiếu. 24% bệnh nhân có thể mắc bệnh tâm thần phân liệt (người lớn). Tuổi thọ có thể bị giảm.

2.2 Các xét nghiệm cận lâm sàng
- Tại thời điểm chẩn đoán
+ Công thức máu toàn phần
+ Imunoglobulin
+ Calci máu, chức năng tuyến giáp
+ Siêu âm tim, điện tim
+ Kiểm tra mất đoạn 22q11 ở bố mẹ hoặc anh/chị/em ruột (kĩ thuật FISH)
+ Siêu âm thận (thận đơn, nang thận, giãn hệ thống ống góp)
+ Khám thính lực và mắt
+ Kiểm tra vẹo cột sống khi chẩn đoán và giai đoạn sớm của tuổi vị thành niên
+ Theo dõi chiều cao và cân nặng thường xuyên đến 2 tuổi, và hàng năm sau đó. Các xét nghiệm liên quan đến chậm tăng trưởng cần được đánh giá đầy đủ, bao gồm cả tầm soát thiếu hụt hormone tăng trưởng.
+ Nhận biết sớm các khó khăn về phát âm vàcan thiệp trị liệu ngôn ngữ nếu cần thiết.
+ Đánh giá tự kỉ, tăng động giảm chú ý và các vấn đề về rối loạn hành vi trongđộ tuổi mầm non và học đường
- Kiểm tra hàng năm:
+ Công thức máu, canxi máu, và chức năng tuyến giáp
+ Chiều cao và cân nặng
+ Theo dõi bệnh tự miễn; xét nghiệm tự kháng thể theo chỉ định lâm sàng
3. Chẩn đoán
Chẩn đoán hội chứng DiGeorge dựa trên việc xác định mất đoạn 22q11.2, đoạn mất có thể từ 1,5Mb đến 3,0Mb, tuy nhiên không có sự khác biệt về kiểu hình có thể nhận biết được giữa các kiểu mất đoạn khác nhau.
Ngày nay với sự phát triển của các phương pháp chẩn đoán về di truyền học đã làm tăng khả năng chẩn đoán hội chứng mất đoạn nhỏ phổ biến nhất này, tuy nhiên, do sự không đồng nhất trên lâm sàng của hội chứng này đã cản trở hay trì hoãn việc chẩn đoán sớm.
Vào giữa những năm 1990, kỹ thuật lai huỳnh quang tại chỗ (FISH) sử dụng một đầu dò đặc trưng cho vùng nhiễm sắc thể 22q11.2 đã được mô tả và trong nhiều năm sau đã được coi là ―tiêu chuẩn vàng‖ để phát hiện ra mất đoạn nhỏ này.Đây cũng là phương pháp phổ biến nhất. Tuy nhiên, kết quả FISH âm tính không loại trừ hoàn toàn chẩn đoán hội chứng mất đoạn 22q11.2.
Ngày nay, các công nghệ trong sinh học phân tử đã cho phép phát triển các phương pháp khác nhau, chẳng hạn như kỹ thuật MLPA, một phương pháp hiệu quả, nhanh chóng và độ nhạy cao để chẩn đoán hội chứng mất đoạn 22q11.2,có thể phát hiện cả các mất đoạn hoặc lặp đoạn nhỏ ở vùng này, cũng như các thay đổi ở các vùng nhiễm sắc thể khác có liên quan đến kiểu hình của hội chứng này.
Gần đây, phương pháp giải trình tự toàn bộ bộ hệ gen đã đạt được thành quả đáng kể trong lĩnh vực di truyền học, và hiện tại, kĩ thuật CMA (choromosomal microarray) là kỹ thuật tốt nhất để khảo sát sự mất đoạn 22q11.2.
4. Điều trị và tiên lượng
Điều trị hội chứng DiGeorge cần phải phối hợp nhiều chuyên khoa. Các tổn thương hay bệnh lý cần phải được thăm khám, đánh giá và điều trị theo các chuyên khoa riêng biệt. Tuy nhiên có một số vấn đề thường gặp và biện pháp xử lí được đề cập trong bảng sau:




TÀI LIỆU THAM KHẢO
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(2):332-9 e1.
- Habel A, Herriot R, Kumararatne D, Allgrove J, Baker K, Baxendale H, et al. Towards a safety net for management of 22q11.2 deletion syndrome: guidelines for our times. Eur J Pediatr. 2014;173(6):757-65.
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071.
- Sgardioli IC, Paoli Monteiro F, Fanti P, Paiva Vieira T, Gil-da-Silva-Lopes VL. Testing criteria for 22q11.2 deletion syndrome: preliminary results of a low cost strategy for public health. Orphanet J Rare Dis. 2019;14(1):123.
"Hướng dẫn chẩn đoán và điều trị bệnh trẻ em (cập nhật 2020)", BV Nhi Trung Ương
Anh chị có thể inbox Pan Happy hoặc liên hệ qua hotline zalo 0964821468 để được hướng dẫn dinh dưỡng cho mẹ và bé khỏe nha ạ.










